Caloric and isothermal equations of state of solids: empirical modeling with multiply broken power-law densities
Abstract
Empirical equations of state (EoSs) are developed for solids, applicable over extended temperature and pressure ranges. The EoSs are modeled as multiply broken power laws, in closed form without the use of ascending series expansions; their general analytic structure is explained and specific examples are studied. The caloric EoS is put to test with two carbon allotropes, diamond and graphite, as well as vitreous silica. To this end, least-squares fits of broken power-law densities are performed to heat capacity data covering several logarithmic decades in temperature, the high- and low-temperature regimes and especially the intermediate temperature range where the Debye theory is of limited accuracy. The analytic fits of the heat capacities are then temperature integrated to obtain the entropy and caloric EoS, i.e. the internal energy. Multiply broken power laws are also employed to model the isothermal EoSs of metals (Al, Cu, Mo, Ta, Au, W, Pt) at ambient temperature, over a pressure range up to several hundred GPa. In the case of copper, the empirical pressure range is extended into the TPa interval with data points from DFT calculations. For each metal, the parameters defining the isothermal EoS (i.e. the density–pressure relation) are inferred by nonlinear regression. The analytic pressure dependence of the compression modulus of each metal is obtained as well, over the full data range.
Keywords
Multi-parameter equation of state (EoS) Caloric EoS of carbon allotropes Specific heat of vitreous silica Thermal EoS and compression modulus of metals High-pressure regime Multiply broken power laws1 Introduction
The aim of this paper is to develop analytic equations of state (EoSs) for solids which can reproduce empirical data sets covering several orders in temperature and pressure, including the extended crossovers between the asymptotic low and high pressure and temperature regimes. The proposed EoSs are multiply broken power laws, which do not involve truncated series expansions in density, pressure or temperature (frequently used in empirical EoSs, cf. e.g. the reviews [1, 2, 3, 4, 5]) and are, therefore, equally suitable for the mentioned asymptotic regions.
The isochoric heat capacities of diamond, graphite and vitreous , discussed in Sect. 2, are special cases of the broken power-law density (1). In the case of diamond, interpolates between the constant Dulong–Petit high-temperature limit and the Debye low-temperature slope. In the intermediate temperature range, the broken power law (1) is sufficiently adaptable to model deviations from the Debye theory such as the boson peaks of glasses. In the case of graphite, interpolates between the linear low-temperature slope of the degenerate electron gas and the classical Dulong–Petit regime, and in the case of the glass, the linear low-temperature slope is replaced by a power law with non-integer index close to one, generated by a fermionic two-level system. In the latter two examples, a phononic cubic temperature slope does not emerge in the empirical data sets, as the low-temperature regime is dominated by Fermi systems.
In Sect. 3, the EoS (2) will be put to test by performing least-squares fits to high-pressure data sets of Al, Cu, Mo, Ta, Au, W, and Pt, which cover an extended pressure range, from ambient (zero) pressure to several hundred GPa [25, 26, 27]. In the case of copper, the available experimental pressure range (up to 450 GPa [26]) will be extended with data sets obtained from DFT calculations [28]. The least-squares fit of the EoS of copper then covers pressures reaching 60 TPa. In Sect. 4, we present our conclusions.
2 Heat capacity, caloric EoS, and entropy of diamond, graphite and vitreous silica
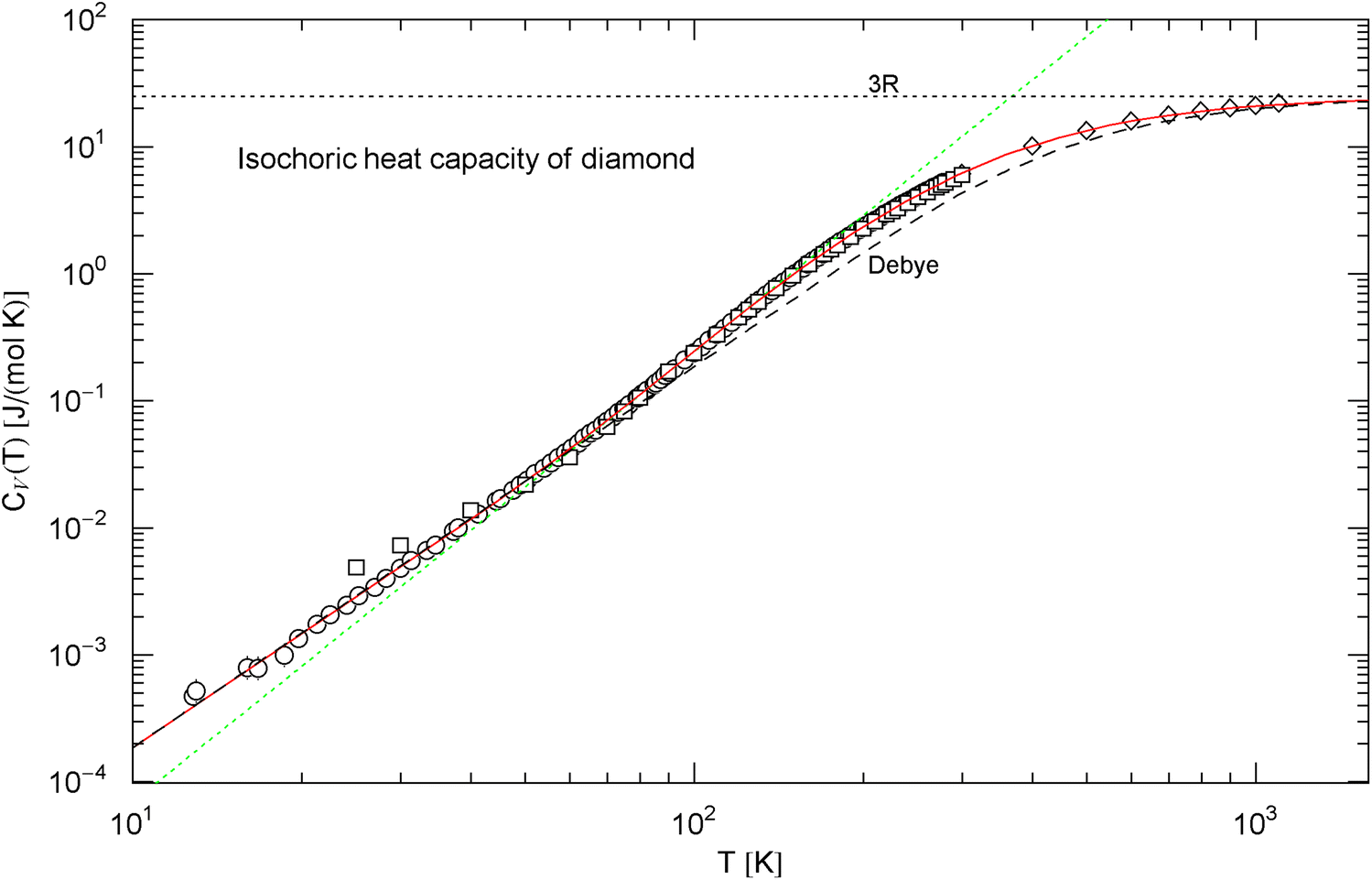
Isochoric heat capacity of diamond, cf. Sect. 2.1. Data points from Ref. [33] (circles), Ref. [34] (squares) and Ref. [35] (diamonds). The fit (solid red curve) is performed with in (3), the fitting parameters are recorded in Table 1. For comparison, the black dashed curve is the Debye approximation (4), with constant Debye temperature of . The classical Dulong–Petit limit is indicated by the black dotted line. The green dotted straight line depicts the tangent at the inflection point (, ), with slope and amplitude
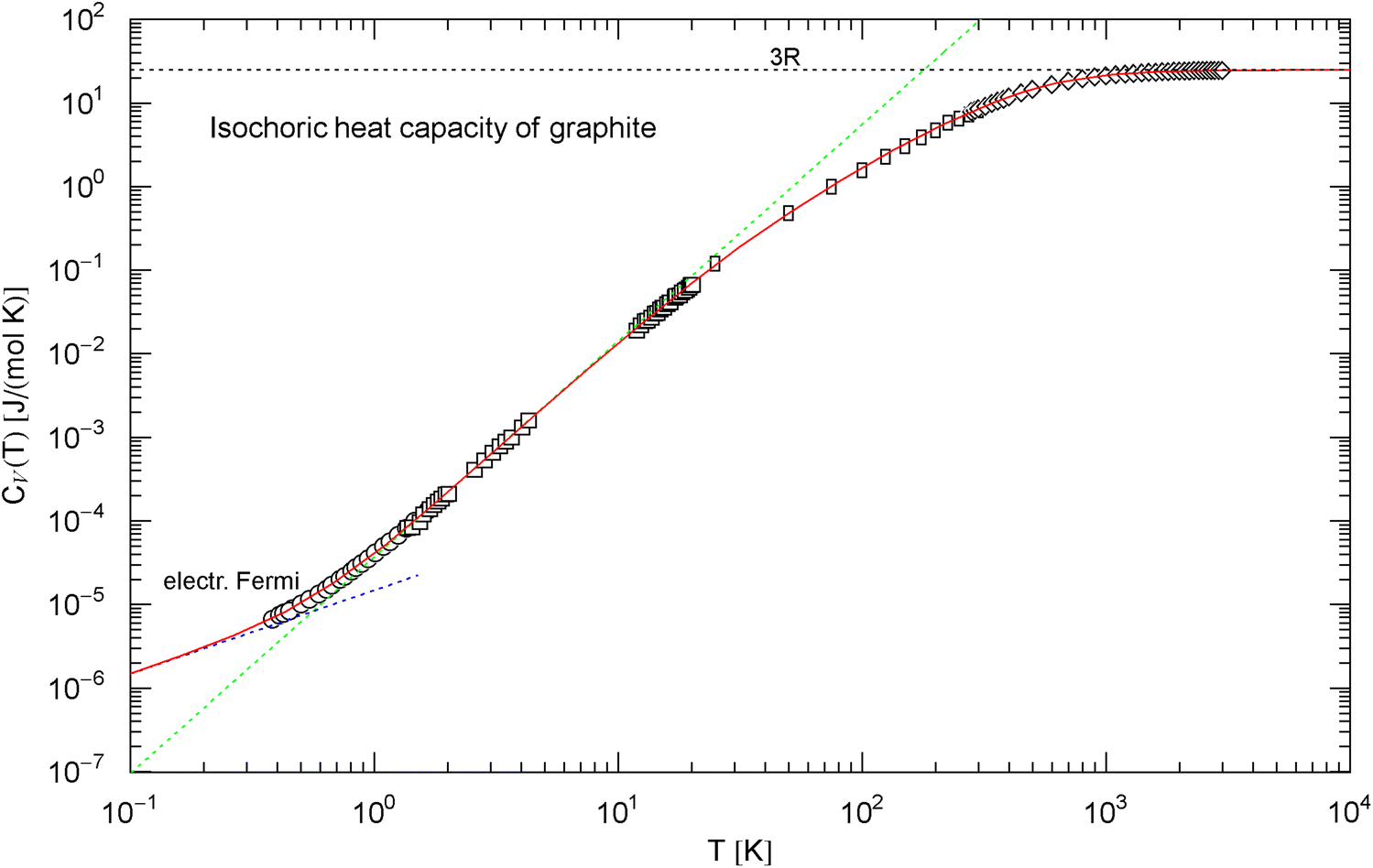
Isochoric heat capacity of graphite, cf. Section 2.2. Data points from Ref. [36] (circles, Madagascar natural graphite), Ref. [37] (squares, Canadian natural graphite), Ref. [38] (rectangles, Acheson graphite), and Ref. [39] (diamonds, Acheson graphite). The experimental isobaric data points have been converted into isochoric points, cf. (6). The fit (red solid curve) is performed with the analytic broken power law in (7), the fitting parameters are listed in Table 1. The dotted blue asymptote is the low-temperature limit of the heat capacity of the electron plasma. The curve has an inflection point at , . The dotted green line is the tangent at the inflection point, with amplitude and slope , which is the maximum slope attained. See also Fig. 4 for the Debye approximation
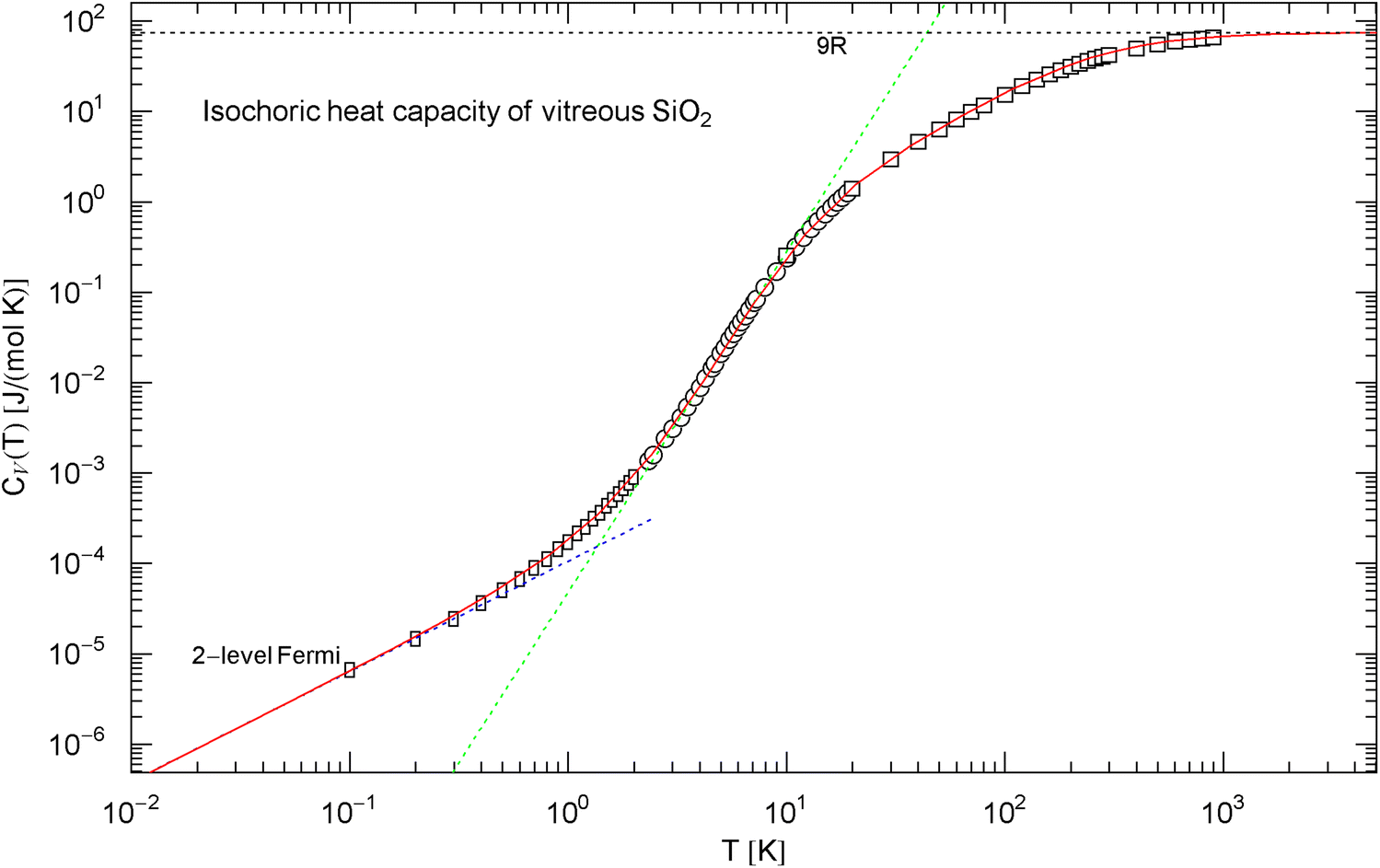
Isochoric heat capacity of , cf. Section 2.3. Data points from Ref. [45] (rectangles), Ref. [46] (circles), and Ref. [47] (squares). The least-squares fit (red solid curve) is performed with the broken power law in (8), the fitting parameters are recorded in Table 1. The classical limit is indicated by the black dotted line. The dotted blue low-temperature asymptote depicts the heat capacity of the two-level Fermi system discussed in Sect. 2.3, which dominates the phononic heat capacity at low temperature. The tangent at the inflection point (, ) has slope and amplitude
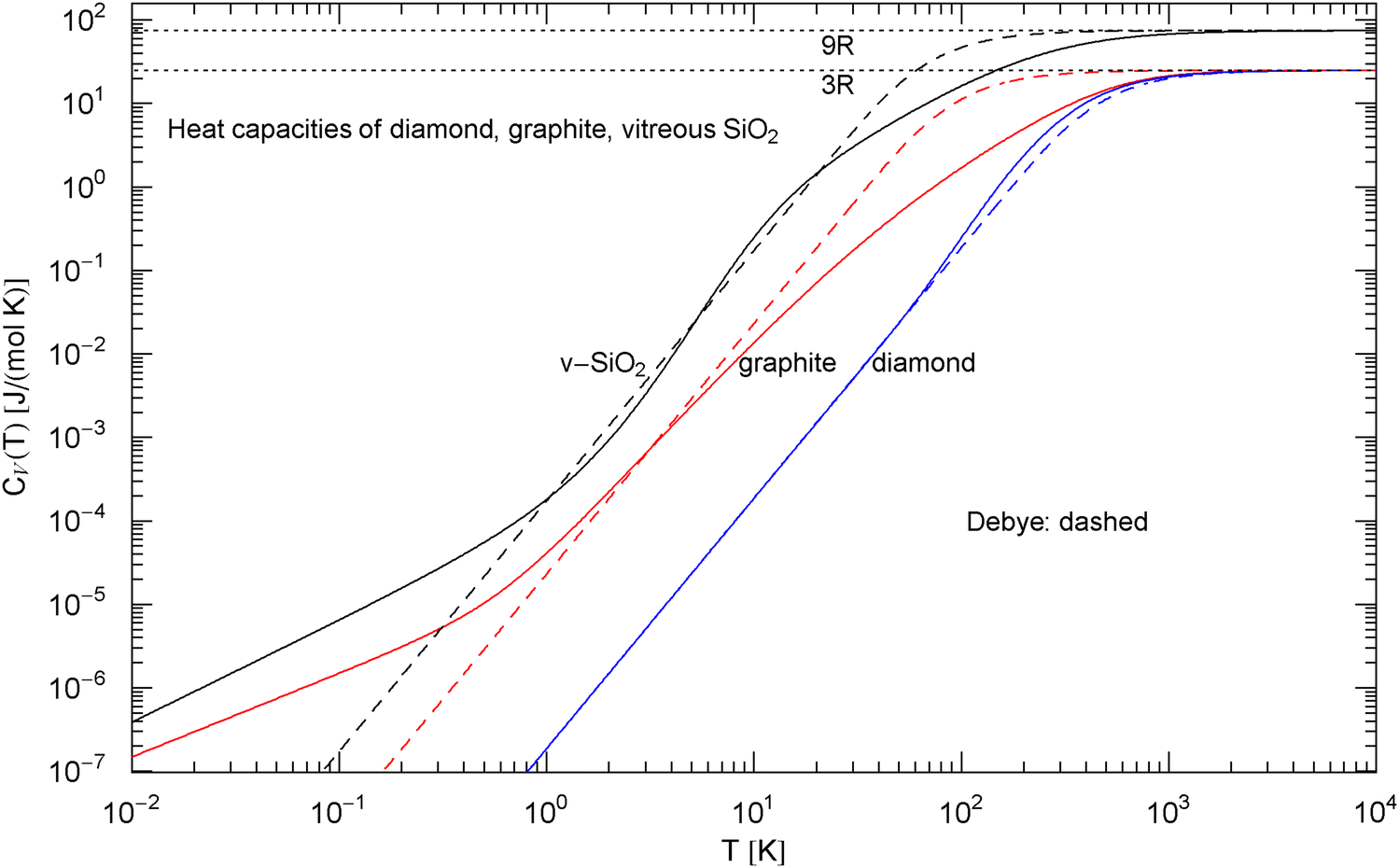
Comparison of the molar isochoric heat capacities of diamond, graphite and vitreous silica. The blue, red and black solid curves show the broken power laws in (3), (7) and (8), respectively, with fitting parameters in Table 1. (The fits are depicted in Figs. 1, 2 and 3). The dashed curves are the Debye heat capacities, cf. (4). The Debye temperature of diamond is (, cf. (4) and Table 1), inferred from the low-temperature limit of the broken power law (3). The Debye temperatures and have been chosen so that the Debye heat capacity (4) intersects the fitted curves at their inflection point, since the empirical heat capacities of graphite and do not exhibit a slope in any temperature range
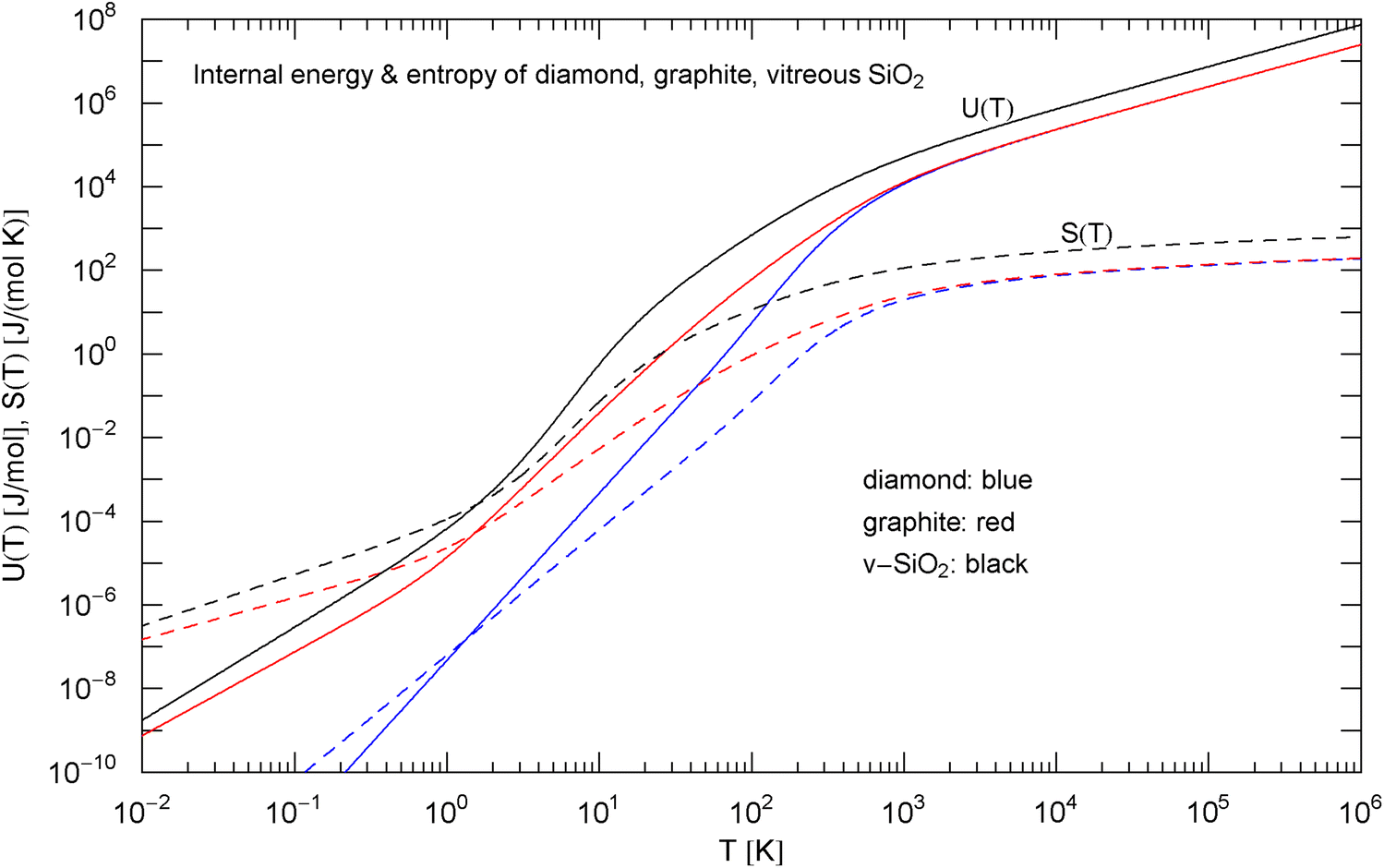
Caloric EoS and molar entropy of diamond, graphite and vitreous silica, cf. (5). The caloric EoSs (thermal component of the internal energy) are depicted as solid curves, the entropies as dashed ones. At high temperature, and the entropy diverges logarithmically. The isochoric heat capacities in the integrands are defined in (3), (7) and (8), with fitting parameters in Table 1. The low-temperature slopes read and , with exponent in Table 1
2.1 Diamond
Diamond | Graphite | ||
|---|---|---|---|
67.435 | 0.60277 | 2.6677 | |
282.02 | 40.466 | 10.585 | |
– | 562.35 | 247.30 | |
3 | 1 | 1.22 | |
1.2496 | 1.6796 | 4.3172 | |
1.3235 | 4.0656 | ||
– | |||
0.30536 | 0.69130 | 2.7558 | |
2.1816 | 1.1347 | 1.7243 | |
– | 0.56727 | 0.93528 | |
0.144 | 0.0539 | 0.0635 | |
dof | 117–5 | 114–8 | 78–8 |
SE | 0.070 | 0.070 | 0.431 |
The asymptotic limits of the Debye function are and , so that and , where denotes the number of atoms per molecule. The units are and as above. The amplitude is taken from the least-squares fit of in (3), , cf. Table 1, to recover the cubic low-temperature slope. Accordingly, the Debye temperature (4) of diamond is . The low-temperature amplitude is the only adjustable parameter of the Debye heat capacity, so that it is not surprising that the Debye approximation becomes inaccurate in the crossover region.
2.2 Graphite
The units are and . The expansion in the basal plane is negligible compared with . In the case of diamond, cf. Sect. 2.1, the conversion to isochoric heat capacities has already been done in the experimental papers. In the case of vitreous silica, cf. Sect. 2.3, in the temperature range of the available data points.
There is no indication of a slope anywhere to be seen in the data set in Fig. 2. Therefore, we choose the amplitude defining the Debye temperature in a way that the low-temperature slope of the Debye curve [stated in (4)] cuts through the inflection point of the empirical heat capacity , see Fig. 4. This gives a Debye temperature of [with in (4)]. A different choice of would just shift the slope parallel to the depicted slope. It is evident from Fig. 4 that the standard Debye approximation (4) cannot give a reasonable fit to the heat capacity of graphite, irrespective of the choice of , except at very high temperature; see also Ref. [41] and references therein for modifications of the Debye theory regarding graphite. The internal energy and entropy functions of graphite are shown in Fig. 5, obtained by integrating the empirical in (7) (with fitting parameters in Table 1) according to Eq. (5).
2.3 Vitreous
The Debye approximation of the heat capacity is depicted in Fig. 4, cf. (4). As in the case of graphite, there is no slope visible in the measured heat capacity. The Debye temperature [ in (4)] is chosen so that intersects the empirical heat capacity [in (8)] at the inflection point. For comparison, the heat capacities of diamond and graphite, cf. (3), (7) and Table 1, are also indicated in Fig. 4. The molar internal energy and entropy of are depicted in Fig. 5, calculated via (5) and (8).
In Sects. 2.1–2.3, we have studied heat capacities and caloric EoSs obtained by fitting broken power laws (1) to empirical data. In the next section, we will use broken power-law densities to model isothermal EoSs of solids at high pressure, as indicated in (2).
3 Isothermal EoS of metals at high pressure
We start with the Murnaghan EoS , cf. e.g. Refs. [19, 51, 52], where denotes the bulk modulus at zero pressure and its pressure derivative at (practically ambient pressure). denotes the mass density and the mass density at zero pressure. The temperature dependence of , and of the constants , , will not be explicitly indicated; the data sets studied here refer to a constant ambient temperature of . Inverting the above EoS, we find and the compressibility , which gives a linear pressure dependence of the compression modulus . Accordingly, and . This linear relation and the Murnaghan EoS are usually applicable up to pressures of a few , cf. e.g. Ref. [53].
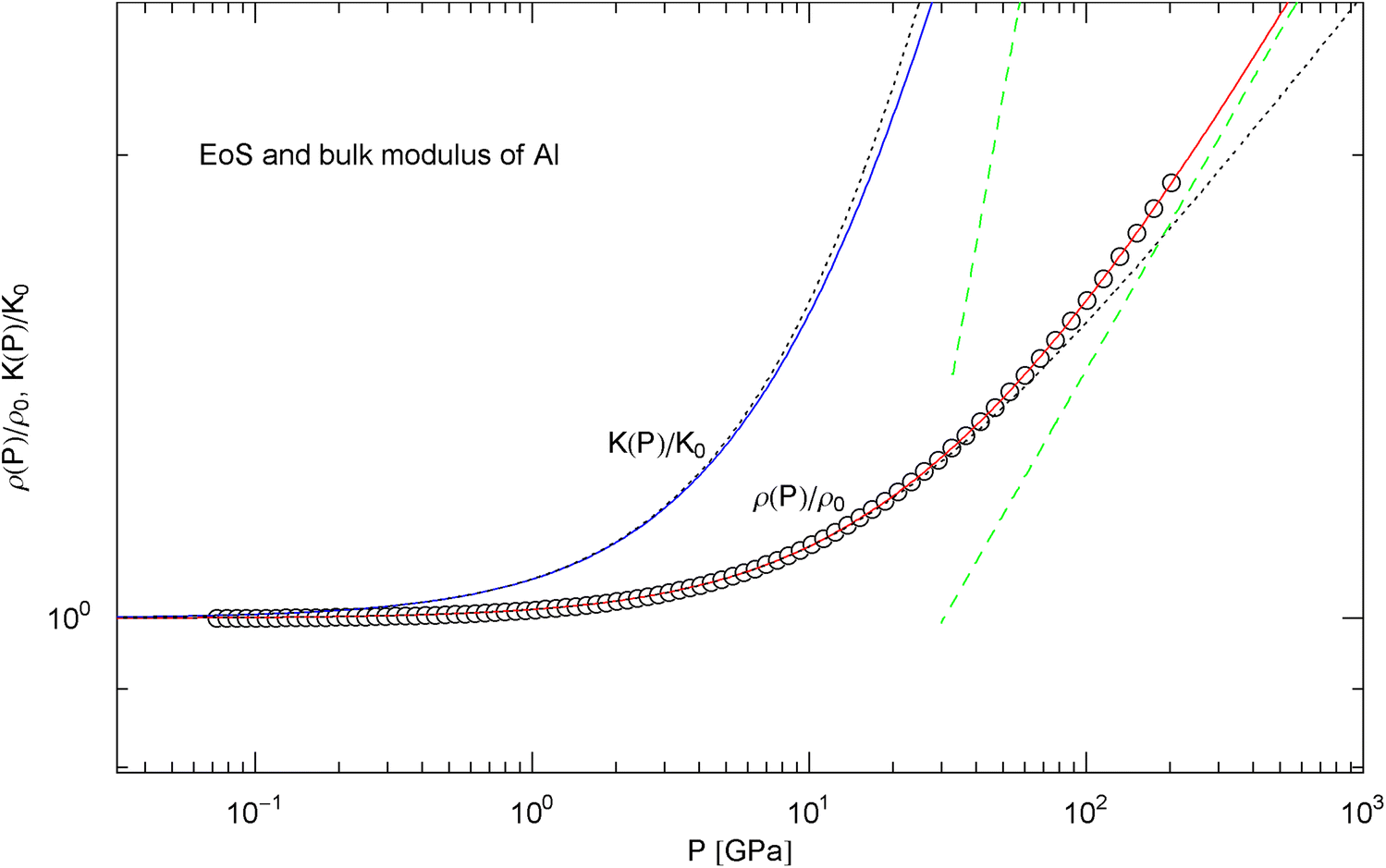
Pressure isotherm at and compression modulus of aluminum. Data points up to 200 GPa (by Hugoniot shock compression) from Ref. [25]. The fit (solid red curve) is performed with the isothermal EoS in (15) (), the fitting parameters , are recorded in Table 2. The solid blue curve depicts the compression modulus calculated from the fitted EoS, cf. (16). and denote mass density and compression modulus at zero pressure, cf. Table 2. The dashed green lines are asymptotes to and . The dotted black curves depict the Murnaghan approximations of the EoS and compression modulus, cf. after (18), calculated with and its derivative obtained from ultrasonic measurements, cf. Table 2
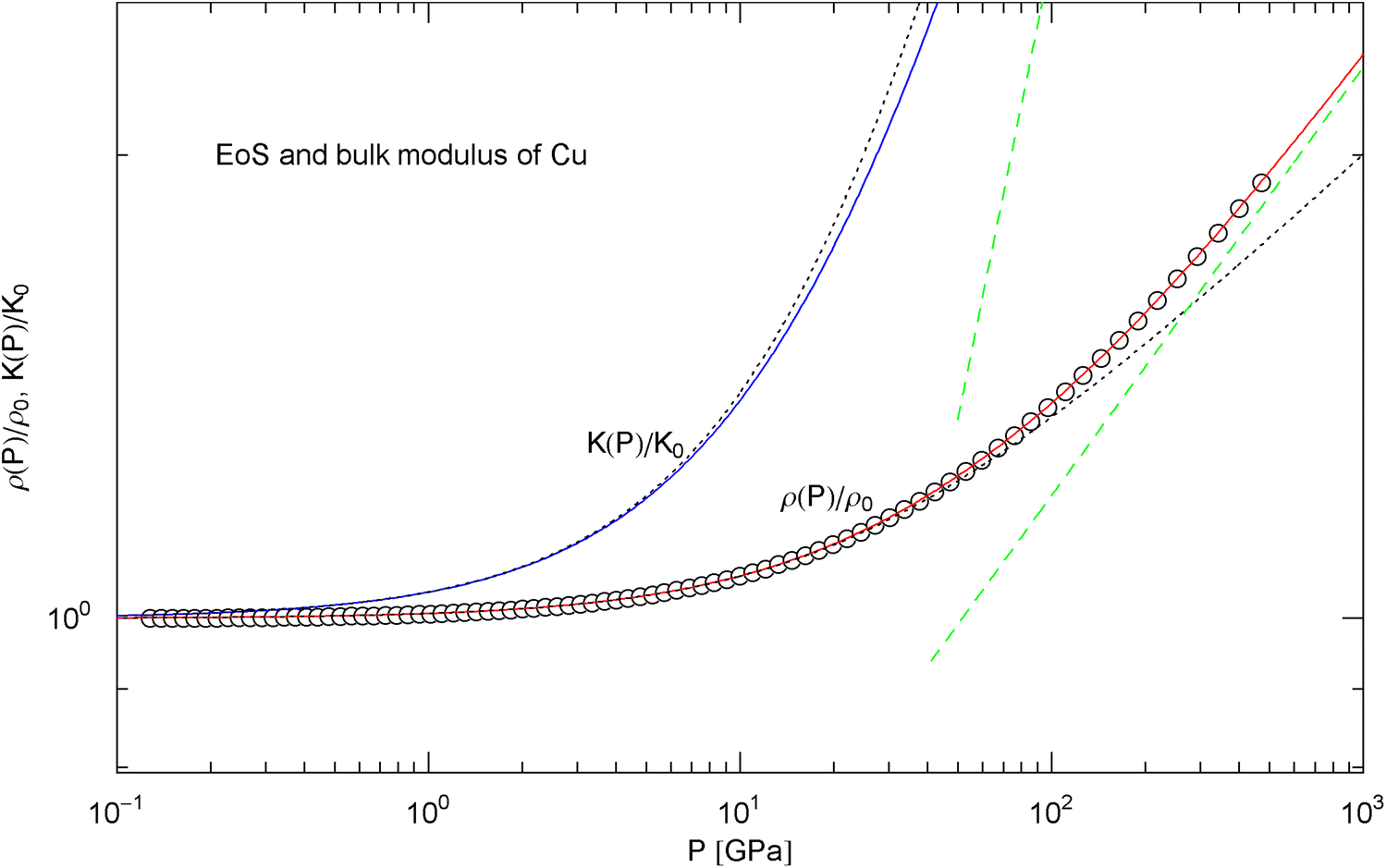
Pressure isotherm and compression modulus of copper. Data points up to 450 GPa (via shockless compression) from Ref. [26]. The caption to Fig. 6 applies. The fit (solid red curve) is performed with EoS in (15), the fitting parameters are listed in Table 2. The solid blue curve shows the compression modulus , cf. (16). The dashed green lines are asymptotes. The dotted black curves depict the Murnaghan approximations, cf. after (18). See also Fig. 13 for the extension of the EoS into the TPa range
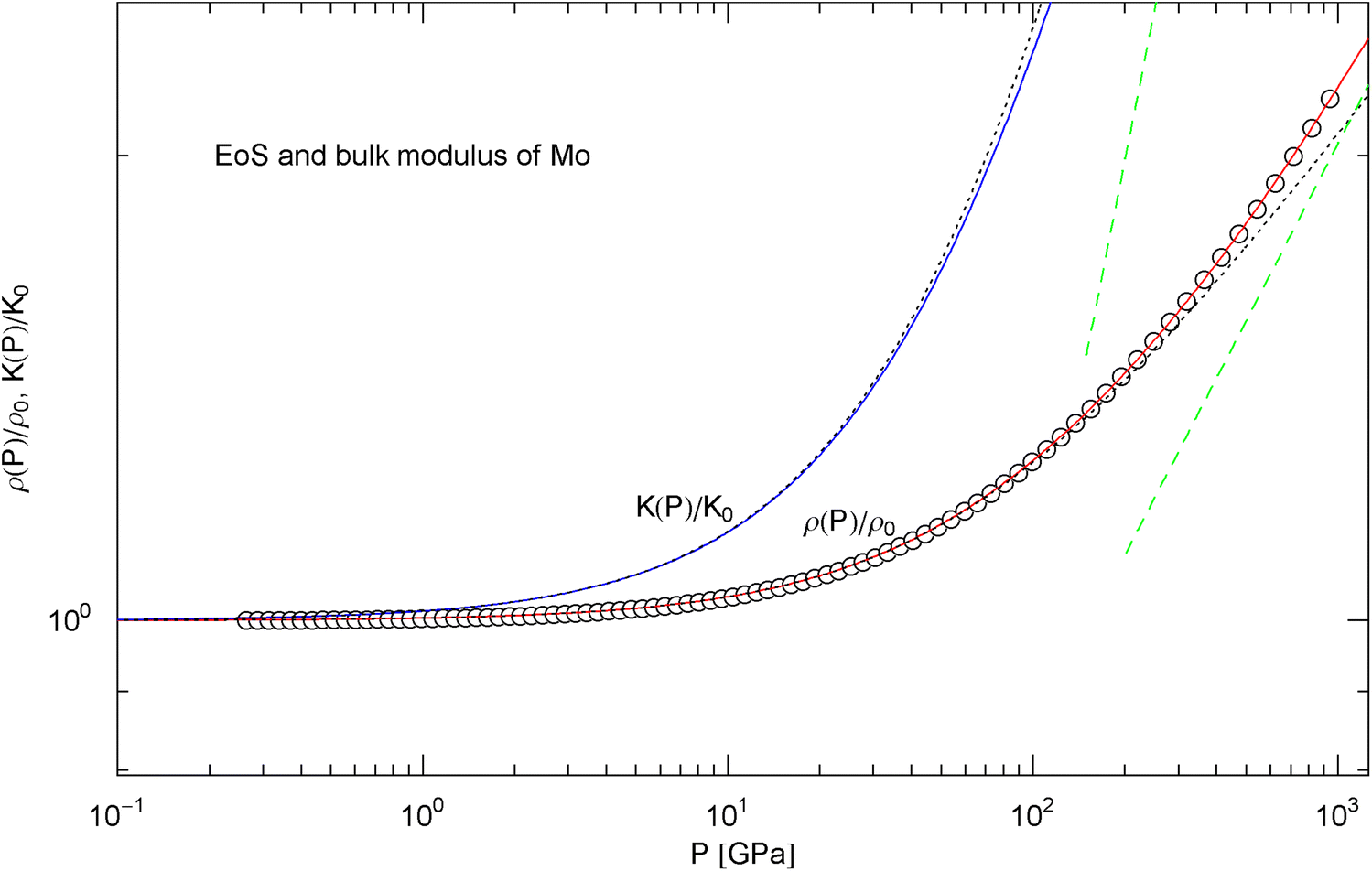
Pressure isotherm and compression modulus of molybdenum. Data points up to 1 TPa (by shock compression) from Ref. [27]. The caption of Fig. 6 applies. The fit (solid red curve) is performed with EoS in (15) and fitting parameters in Table 2. The solid blue curve shows the compression modulus , cf. (16). The dashed green lines are asymptotes and the dotted black curves depict the Murnaghan approximations
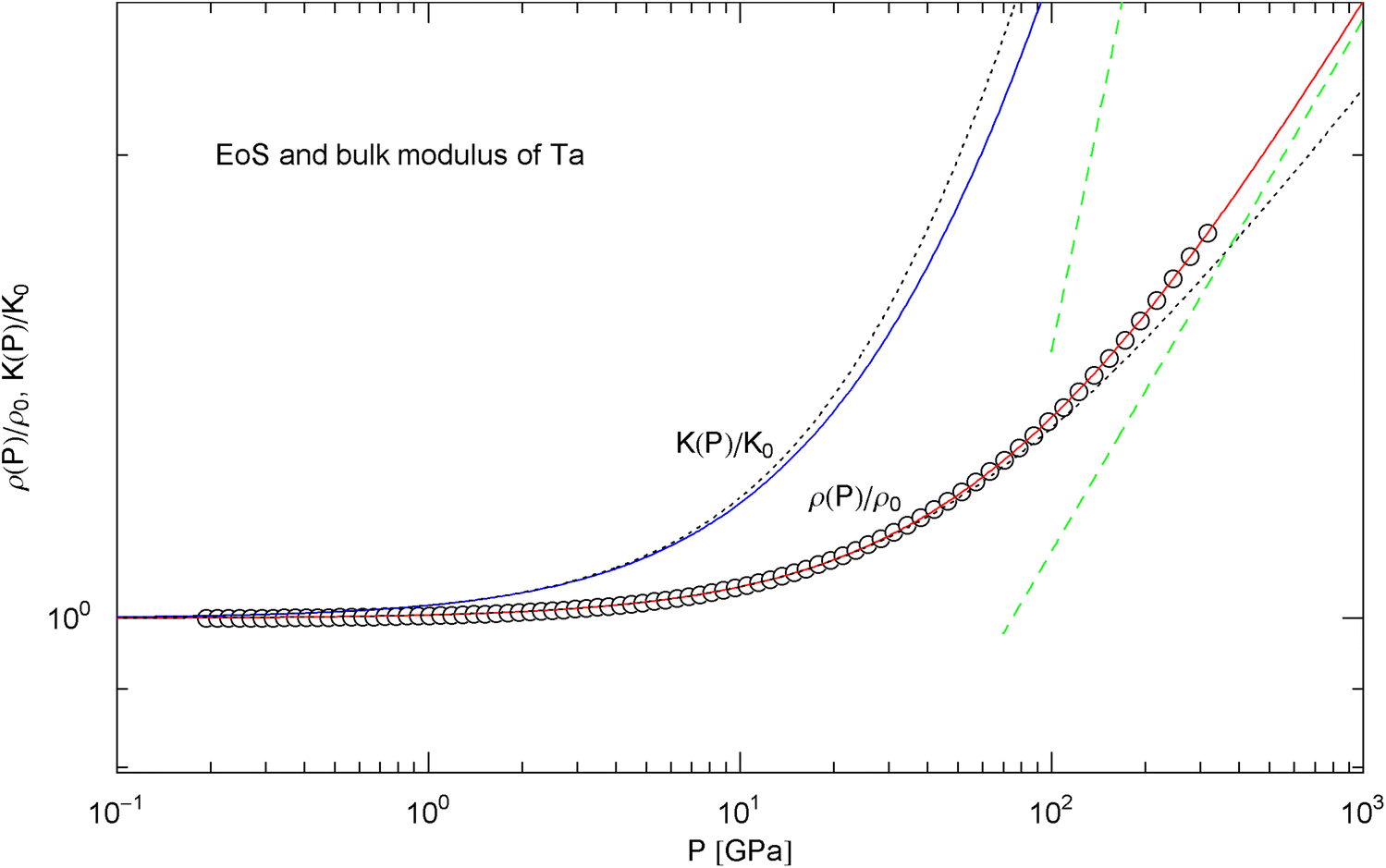
Pressure isotherm and compression modulus of tantalum. Data points up to 300 GPa by shock compression [25]. The fit (solid red curve) is performed with EoS in (15) and fitting parameters in Table 2. The compression modulus , cf. (16), is depicted as blue solid curve. The dashed green lines are the asymptotes of and . The dotted black curves depict the Murnaghan EoS and the linear compression modulus derived from it, cf. after (18)
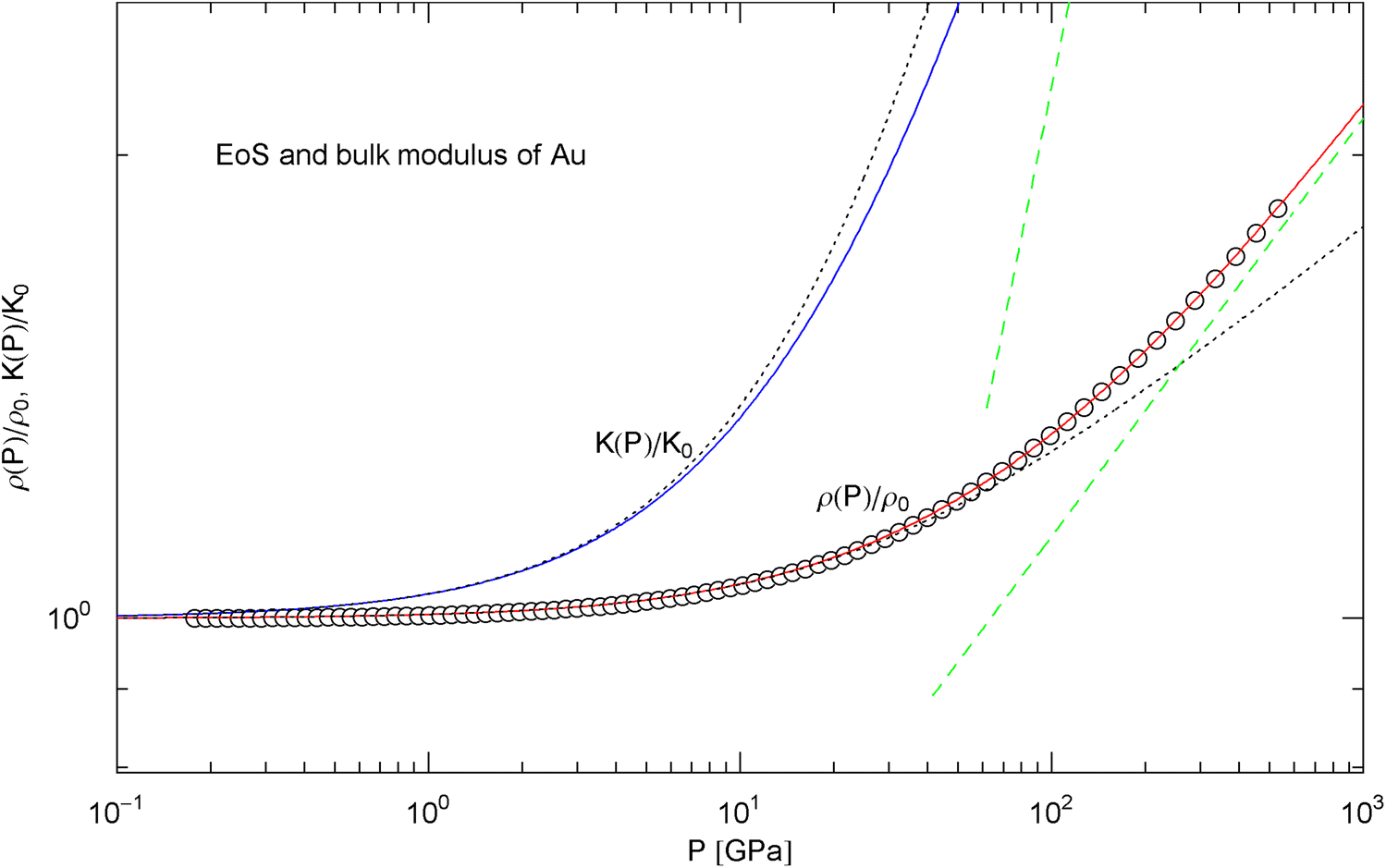
Pressure isotherm at and compression modulus of gold. Data points up to 500 GPa by shock compression [27]. The fit (solid red curve) is performed with EoS in (15), the fitting parameters are recorded in Table 2. The solid blue curve shows the compression modulus , cf. (16). The dashed green lines are asymptotes and the dotted black curves depict the Murnaghan approximations
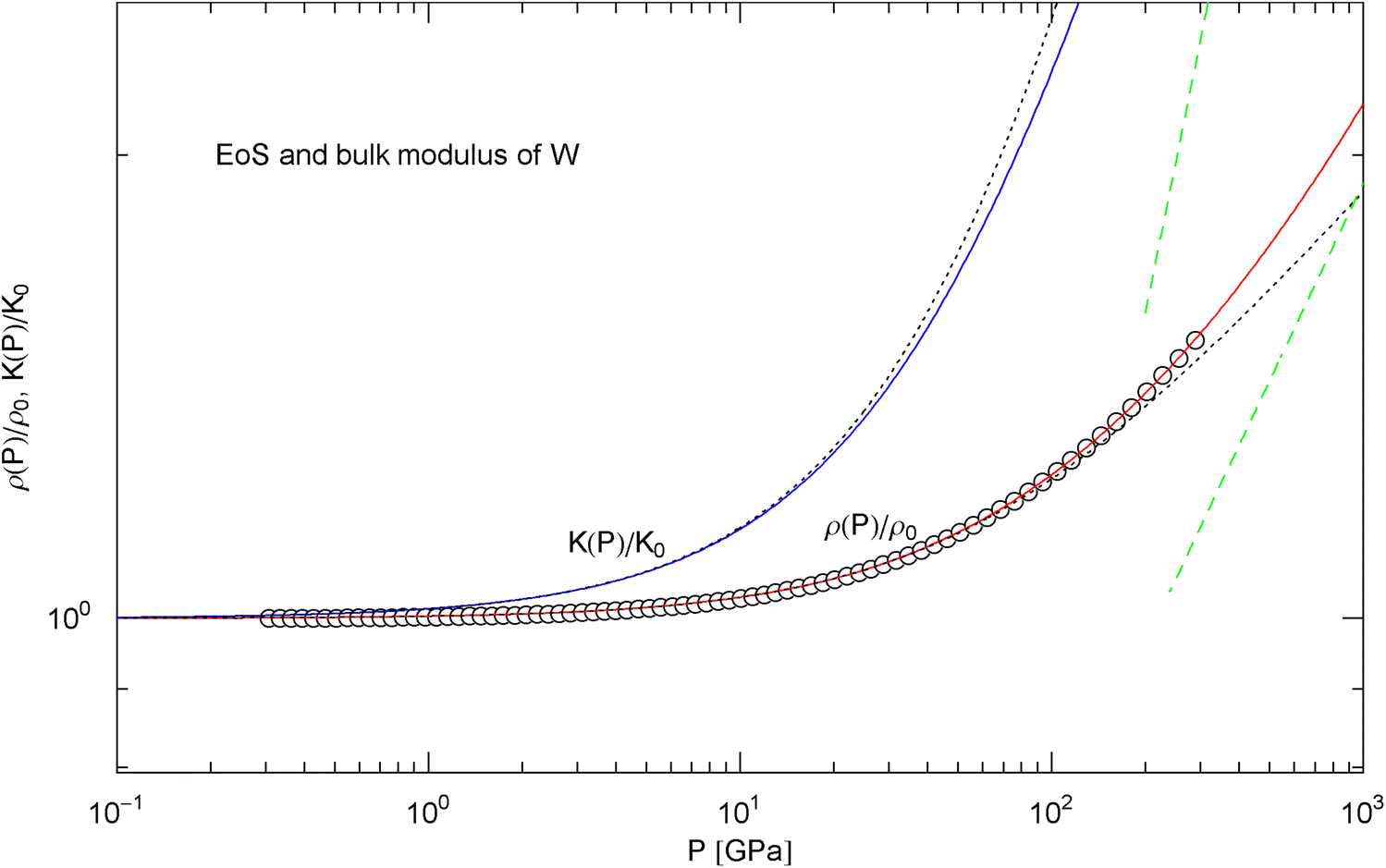
Pressure isotherm at and compression modulus of tungsten. Data points up to 300 GPa by shock compression [25]. The fit (solid red curve) is performed with EoS in (15) and fitting parameters in Table 2. The solid blue curve is the compression modulus , cf. (16). The dotted black curves depict the Murnaghan EoS and the linear approximation of the compression modulus, cf. after (18). The dashed green lines are asymptotes
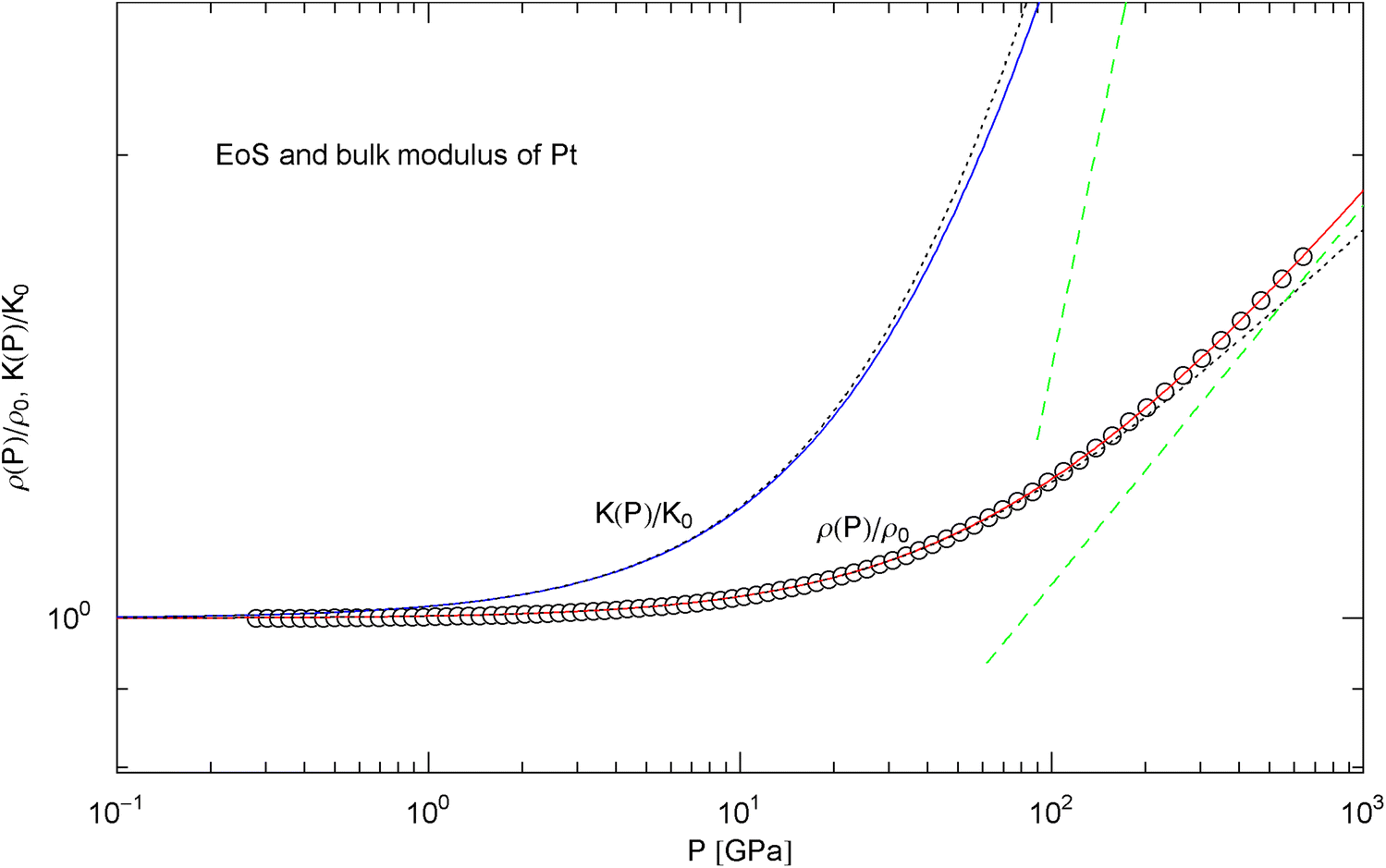
Pressure isotherm and compression modulus of platinum. Data points up to 660 GPa by shock compression [27]. The fit (solid red curve) is performed with EoS in (15), the fitting parameters are recorded in Table 2. The solid blue curve shows the compression modulus , cf. (16). The dashed green lines are asymptotes and the dotted black curves depict the Murnaghan approximations, cf. after (18)
Fitting parameters of the pressure isotherms of Al, Cu, Mo, Ta, Au, W and Pt, at ambient temperature of . The isothermal EoS used for the fits is defined in (15)
Al | Cu | Mo | Ta | Au | W | Pt | Cu @ TPa | |
|---|---|---|---|---|---|---|---|---|
2.707 | 8.939 | 10.22 | 16.67 | 19.24 | 19.25 | 21.41 | 8.939 | |
73 | 133.5 | 264.87 | 194 | 166.7 | 296 | 280.03 | 133.5 | |
4.42 | 5.36 | 3.7499 | 3.83 | 6.23 | 4.3 | 5.0886 | 5.36 | |
78.084 | 120.03 | 612.12 | 90.405 | 114.00 | 531.46 | 169.13 | 113.31 | |
0.13699 | 0.14094 | 0.14521 | 0.28648 | 0.17773 | 0.25268 | 0.10226 | 0.13916 | |
– | – | – | – | – | – | – | 6507.1 | |
– | – | – | – | – | – | – | 0.15077 | |
83.723 | 158.32 | 282.63 | 503.58 | 225.24 | 344.48 | 337.11 | 160.28 | |
5.6564 | 7.2929 | 4.2386 | 16.918 | 10.680 | 5.7177 | 6.9681 | 7.4471 | |
dof | 82 | 82 | 84 | 80 | 81 | 75 | 78 | 112 |
SE | ||||||||
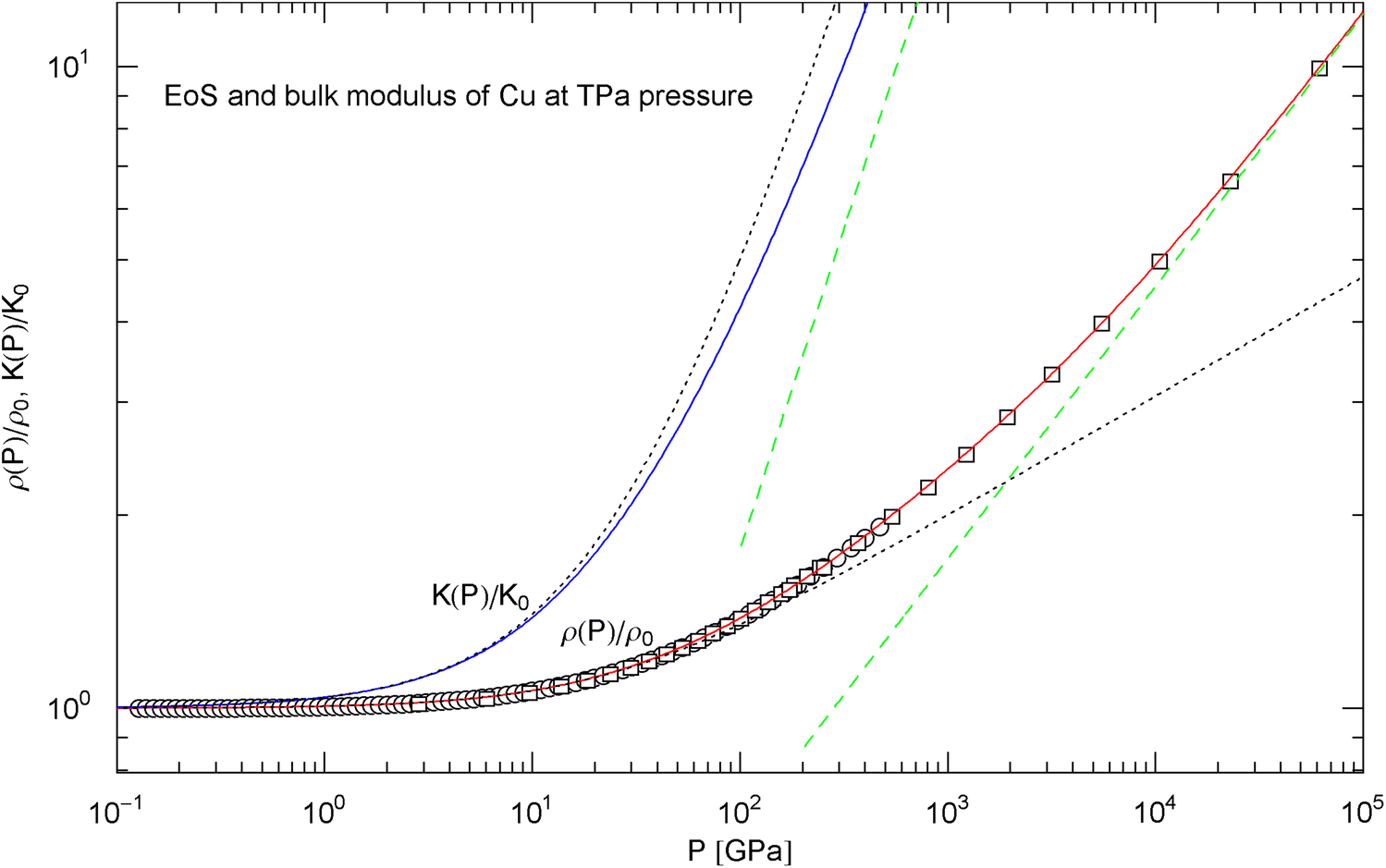
Pressure isotherm at and compression modulus of copper up to 60 TPa. Data points up to 450 GPa (circles) obtained by shockless compression [26] and up to 60 TPa (squares) from DFT calculations [28]. The fit (solid red curve) is performed with the isothermal EoS in (15) (with ), the fitting parameters are listed in Table 2. The solid blue curve shows the compression modulus , cf. (16). The dotted black curves depict the Murnaghan EoS and the linear compression modulus derived from it, cf. after (18). The dashed green straight lines indicate the asymptotes of the pressure isotherm and compression modulus
In Figs. 6, 7, 8, 9, 10, 11, 12 and 13, we have also depicted the pressure dependence of the compression modulus (solid blue curves) defined in (16), using input and fitting parameters of the regressed EoS (15) listed in Table 2. In these figures, we also compare the EoS (15) with the Murnaghan EoS outlined at the beginning of this section [we write here and for density and density at zero pressure to distinguish this EoS from the general EoS in (15)]. The compression modulus in (16) is also compared with the linear relation obtained from the Murnaghan EoS. (The linearity of is somewhat hidden in the log–log representation of the figures.) Actually, this linear relation, experimentally verified up to pressures of a few GPa (but usually invalid above 10 GPa, see Figs. 6, 7, 8, 9, 10, 11 and 12) is the starting point when deriving the Murnaghan EoS, by substituting and integrating.
The EoS of a non-relativistic degenerate electron gas in the Thomas–Fermi free-electron approximation gives [51, 66], leading to the condition to be satisfied by the exponents and in (15) and (20). Application of this relation to the EoS of copper depicted in Fig. 13 (which is accurate up to 60 TPa and based on EoS (15) with and parameters in Table 2) suggests that an additional factor with will be necessary in EoS (15) () to extend the density–pressure curve beyond 60 TPa. (In the case of an ultra-relativistic degenerate electron plasma, is replaced by , so that .) Phenomenological thermodynamic arguments were used in Refs. [67, 68] to suggest the inequality instead of . In contrast to the positivity of the heat capacities and compressibilities, this is not a necessary equilibrium condition but presumably more realistic for planetary cores than the Thomas–Fermi limit [5, 69]. The inequality weakens the above condition to and gives the constraint on the exponent of the fourth factor in the ultra-high pressure EoS of copper applicable beyond 60 TPa.
4 Conclusion
The examples discussed in Sects. 2 and 3 demonstrate the efficiency of multiply broken power laws in modeling empirical equations of state of solids. The heat capacities and mass densities defined in Sect. 1 are analytic functions composed of power-law segments joint by smooth analytic transitions; they do not involve series expansions, being assembled as products of power-law factors , , cf. (1) and (2), where stands for the temperature or pressure variable. Broken power laws are particularly suitable to model data sets where pressure or temperature varies over several orders of magnitude, interpolating between different asymptotic regimes. The condition on the amplitudes defining the power-law factors allows us to systematically assemble caloric and isothermal EoSs over an ever increasing temperature or pressure range; since , the factor with amplitude does not significantly affect the EoS in the range .
In Sect. 2, we discussed three examples of caloric EoSs (of two carbon allotropes and vitreous ); the corresponding isochoric heat capacities exhibit very different low-temperature power-law scaling and also deviate noticeably from the Debye theory in the intermediate temperature range. The log–log plots in Figs. 1, 2 and 3 depict least-squares fits of the heat capacities [modeled as broken power laws, cf. (3), (7) and (8)] to the respective isochoric data sets of diamond, graphite and . For these materials, heat capacity measurements (including the conversion from isobaric to isochoric heat capacities by way of Grüneisen parameter and thermal expansion coefficient) are available over an extended temperature range covering the low- and high-temperature regimes. As these examples demonstrate, broken power laws are sufficiently adaptable to accurately reproduce the heat capacities over the full empirical temperature range, including features such as the boson peak of the glass in Fig. 4 emerging at around 10 K or the excess heat capacity (as compared to the Debye theory) of diamond peaking at 200 K, cf. Figs. 1 and 4. The analytic heat capacities obtained from the least-squares fits can be temperature integrated to obtain the caloric EoS and entropy variable, see Fig. 5.
In Sect. 3, we discussed isothermal EoSs of specific metals, capable of reproducing high-pressure data sets. The examples studied cover a wide range of mass densities (at ambient pressure), from Al to Pt, cf. Table 2. In contrast to the multiply broken power laws modeling the heat capacities in Sect. 2, which are non-analytic at , we used broken power laws analytic at zero pressure, cf. (2). The EoS can then be made to converge to the Murnaghan EoS in the low-pressure regime, where the compression modulus is known to vary linearly with pressure. The least-squares fits of the isothermal EoS (15) to high-pressure data sets of Al, Cu, Mo, Ta, Au, W and Pt are depicted in Figs. 6, 7, 8, 9, 10, 11 and 12. In all cases, the fits are uniformly accurate over the full empirical pressure range, from ambient pressure to several hundred GPa. In Fig. 13, the empirical EoS of Cu is extended into the TPa regime, using data points from DFT calculations for the least-squares regression. In Sect. 3, we also explained the implementation of the ultra-high pressure Thomas–Fermi limit into empirical isothermal EoSs defined by multiply broken power laws.
Notes
References
- 1.W.B. Holzapfel, High Press. Res. 16, 81 (1998)ADSCrossRefGoogle Scholar
- 2.W.B. Holzapfel, Z. Kristallogr. 216, 473 (2001)Google Scholar
- 3.J.S. Tse, W.B. Holzapfel, J. Appl. Phys. 104, 043525 (2008)ADSCrossRefGoogle Scholar
- 4.J. Hama, K. Suito, J. Phys. Condens. Matter 8, 67 (1996)ADSCrossRefGoogle Scholar
- 5.F.D. Stacey, Rep. Prog. Phys. 68, 341 (2005)ADSCrossRefGoogle Scholar
- 6.R. Tomaschitz, Physica A (2020). https://doi-org. /10.1016/j.physa.2019.123188 CrossRefGoogle Scholar
- 7.R. Tomaschitz, Physica A 483, 438 (2017)ADSMathSciNetCrossRefGoogle Scholar
- 8.R. Tomaschitz, Fluid Phase Equilib. 496, 80 (2019)CrossRefGoogle Scholar
- 9.K.V. Khishchenko, J. Phys: Conf. Ser. 946, 012082 (2018)Google Scholar
- 10.K.V. Khishchenko, J. Phys. Conf. Ser. 1147, 012001 (2019)CrossRefGoogle Scholar
- 11.K.V. Khishchenko, Tech. Phys. Lett. 30, 829 (2004)ADSCrossRefGoogle Scholar
- 12.D.V. Minakov, P.R. Levashov, K.V. Khishchenko, AIP Conf. Proc. 1426, 836 (2012)ADSCrossRefGoogle Scholar
- 13.D.V. Minakov, P.R. Levashov, K.V. Khishchenko, V.E. Fortov, J. Appl. Phys. 115, 223512 (2014)ADSCrossRefGoogle Scholar
- 14.M.A. Kadatskiy, K.V. Khishchenko, J. Phys: Conf. Ser. 653, 012079 (2015)Google Scholar
- 15.M.A. Kadatskiy, K.V. Khishchenko, J. Phys. Conf. Ser. 774, 012005 (2016)CrossRefGoogle Scholar
- 16.M.A. Kadatskiy, K.V. Khishchenko, Phys. Plasmas 25, 112701 (2018)ADSCrossRefGoogle Scholar
- 17.K.V. Khishchenko, J. Phys. Conf. Ser. 121, 022025 (2008)CrossRefGoogle Scholar
- 18.K.V. Khishchenko, J. Phys. Conf. Ser. 653, 012081 (2015)CrossRefGoogle Scholar
- 19.J.R. Macdonald, Rev. Mod. Phys. 38, 669 (1966)ADSCrossRefGoogle Scholar
- 20.B.G. Yalcin, Appl. Phys. A 122, 456 (2016)ADSCrossRefGoogle Scholar
- 21.S. Khatta, S.K. Tripathi, S. Prakash, Appl. Phys. A 123, 582 (2017)ADSCrossRefGoogle Scholar
- 22.M. Kaddes, K. Omri, N. Kouaydi, M. Zemzemi, Appl. Phys. A 124, 518 (2018)ADSCrossRefGoogle Scholar
- 23.W. Ouerghui, M.S. Alkhalifah, Appl. Phys. A 125, 374 (2019)ADSCrossRefGoogle Scholar
- 24.A. Laroussi, M. Berber, B. Doumi, A. Mokaddem, H. Abid, A. Boudali, H. Bahloul, H. Moujri, Appl. Phys. A 125, 676 (2019)ADSCrossRefGoogle Scholar
- 25.A.D. Chijioke, W.J. Nellis, I.F. Silvera, J. Appl. Phys. 98, 073526 (2005)ADSCrossRefGoogle Scholar
- 26.R.G. Kraus, J.-P. Davis, C.T. Seagle, D.E. Fratanduono, D.C. Swift, J.L. Brown, J.H. Eggert, Phys. Rev. B 93, 134105 (2016)ADSCrossRefGoogle Scholar
- 27.Y. Wang, R. Ahuja, B. Johansson, J. Appl. Phys. 92, 6616 (2002)ADSCrossRefGoogle Scholar
- 28.C.W. Greeff, J.C. Boettger, M.J. Graf, J.D. Johnson, J. Phys. Chem. Solids 67, 2033 (2006)ADSCrossRefGoogle Scholar
- 29.L.E. Fried, W.M. Howard, Phys. Rev. B 61, 8734 (2000)ADSCrossRefGoogle Scholar
- 30.K.V. Khishchenko, V.E. Fortov, I.V. Lomonosov, M.N. Pavlovskii, G.V. Simakov, M.V. Zhernokletov, AIP Conf. Proc. 620, 759 (2002)ADSCrossRefGoogle Scholar
- 31.K.V. Khishchenko, V.E. Fortov, I.V. Lomonosov, Int. J. Thermophys. 26, 479 (2005)ADSCrossRefGoogle Scholar
- 32.S.Sh. Rekhviashvili, Kh.L. Kunizhev, High Temp. 55, 312 (2017)CrossRefGoogle Scholar
- 33.J.E. Desnoyers, J.A. Morrison, Philos. Mag. 3, 42 (1958)ADSCrossRefGoogle Scholar
- 34.W. DeSorbo, J. Chem. Phys. 21, 876 (1953)ADSCrossRefGoogle Scholar
- 35.A.C. Victor, J. Chem. Phys. 36, 1903 (1962)ADSCrossRefGoogle Scholar
- 36.B.J.C. van der Hoeven, P.H. Keesom, Phys. Rev. 130, 1318 (1963)ADSCrossRefGoogle Scholar
- 37.W. DeSorbo, G.E. Nichols, J. Phys. Chem. Solids 6, 352 (1958)ADSCrossRefGoogle Scholar
- 38.W. DeSorbo, W.W. Tyler, J. Chem. Phys. 21, 1660 (1953)ADSCrossRefGoogle Scholar
- 39.M.W. Chase, NIST-JANAF Thermochemical Tables, 4th ed. (AIP, Woodbury, 1998), https://janaf.nist.gov
- 40.A.T.D. Butland, R.J. Maddison, J. Nucl. Mater. 49, 45 (1973)ADSCrossRefGoogle Scholar
- 41.T. Nihira, T. Iwata, Phys. Rev. B 68, 134305 (2003)ADSCrossRefGoogle Scholar
- 42.V.N. Senchenko, R.S. Belikov, J. Phys: Conf. Ser. 891, 012338 (2017)Google Scholar
- 43.J.C. Lasjaunias, A. Ravex, M. Vandorpe, S. Hunklinger, Solid State Commun. 17, 1045 (1975)ADSCrossRefGoogle Scholar
- 44.R.O. Pohl, in: Amorphous Solids, W.A. Phillips, ed. (Springer, Berlin, 1981)Google Scholar
- 45.R.B. Stephens, Phys. Rev. B 8, 2896 (1973)ADSCrossRefGoogle Scholar
- 46.P. Flubacher, A.J. Leadbetter, J.A. Morrison, B.P. Stoicheff, J. Phys. Chem. Solids 12, 53 (1959)ADSCrossRefGoogle Scholar
- 47.R.C. Lord, J.C. Morrow, J. Chem. Phys. 26, 230 (1957)ADSCrossRefGoogle Scholar
- 48.P.W. Anderson, B.I. Halperin, C.M. Varma, Philos. Mag. 25, 1 (1972)ADSCrossRefGoogle Scholar
- 49.W.A. Phillips, Rep. Prog. Phys. 50, 1657 (1987)ADSCrossRefGoogle Scholar
- 50.I.S. Gradshteyn, I.M. Ryzhik, Table of Integrals, Series, and Products, 8th edn. (Academic Press, Waltham, 2015)zbMATHGoogle Scholar
- 51.W.B. Holzapfel, Rep. Prog. Phys. 59, 29 (1996)ADSCrossRefGoogle Scholar
- 52.W.B. Holzapfel, High Press. Res. 22, 209 (2002)ADSCrossRefGoogle Scholar
- 53.G.M. Amulele, M.H. Manghnani, S. Marriappan, X. Hong, F. Li, X. Qin, H.P. Liermann, J. Appl. Phys. 103, 113522 (2008)ADSCrossRefGoogle Scholar
- 54.A. Dewaele, P. Loubeyre, M. Mezouar, Phys. Rev. B 70, 094112 (2004)ADSCrossRefGoogle Scholar
- 55.W.B. Holzapfel, High Press. Res. 30, 372 (2010)ADSCrossRefGoogle Scholar
- 56.K. Katahara, M. Manghnani, E. Fisher, J. Appl. Phys. 47, 434 (1976)ADSCrossRefGoogle Scholar
- 57.K.W. Katahara, M.H. Manghnani, E.S. Fisher, J. Phys. F: Met. Phys. 9, 773 (1979)ADSCrossRefGoogle Scholar
- 58.P. van’t-Klooster, N.J. Trappeniers, S.N. Biswas, Physica B + C 97, 65 (1979)Google Scholar
- 59.S.N. Biswas, P. van’t-Klooster, N.J. Trappeniers, Physica B + C 103, 235 (1981)Google Scholar
- 60.J.L. Tallon, A. Wolfenden, J. Phys. Chem. Solids 40, 831 (1979)ADSCrossRefGoogle Scholar
- 61.D. Steinberg, J. Phys. Chem. Solids 43, 1173 (1982)ADSCrossRefGoogle Scholar
- 62.W. Holzapfel, M. Hartwig, W. Sievers, J. Phys. Chem. Ref. Data 30, 515 (2001)ADSCrossRefGoogle Scholar
- 63.K. Syassen, W.B. Holzapfel, J. Appl. Phys. 49, 4427 (1978)ADSCrossRefGoogle Scholar
- 64.K. Takemura, A. Dewaele, Phys. Rev. B 78, 104119 (2008)ADSCrossRefGoogle Scholar
- 65.W.B. Holzapfel, M.F. Nicol, High Press. Res. 27, 377 (2007)ADSCrossRefGoogle Scholar
- 66.E.E. Salpeter, Astrophys. J. 134, 669 (1961)ADSMathSciNetCrossRefGoogle Scholar
- 67.F.D. Stacey, Geophys. J. Int. 143, 621 (2000)ADSCrossRefGoogle Scholar
- 68.F.D. Stacey, P.M. Davis, Phys. Earth Planet. Inter. 142, 137 (2004)ADSCrossRefGoogle Scholar
- 69.F.D. Stacey, J.H. Hodgkinson, Phys. Earth Planet. Inter. 286, 42 (2019)ADSCrossRefGoogle Scholar